FLAIR modules¶
flair.py is a wrapper script with modules for running various
processing scripts located in bin/. Modules are assumed to be run in
order (align, correct, collapse), but the user can forgo the wrapper if
a more custom build is desired.
flair align¶
Aligns reads to the genome using minimap2, and converts the aligned
minimap2 sam output to
BED12 and
optionally PSL.
Aligned reads in psl format can be visualized in IGV or the UCSC
Genome browser. As for which human reference genome to use, Heng Li has
written a blog
post
on this.
Alternatively, the user can align the reads themselves with their
aligner of choice and convert sorted bam output to bed12 using
bin/bam2Bed12.py to supply for flair-correct. This step smooths gaps
in the alignment.
Usage:
python flair.py align -g genome.fa -r <reads.fq.gz>|<reads.fq>|<reads.fa> [options]
run with --help for a description of optional arguments. Outputs (1)
sam of raw aligned reads and (2) smoothed bed12 file of aligned
reads to be supplied to flair-correct.
flair correct¶
Corrects misaligned splice sites using genome annotations and/or
short-read splice junctions. Based on common user issues we have
encountered, for flair-correct to run properly, please ensure/note that
(1) the genome annotation and genome sequences are compatible, (2)
gtf is preferred over gff for annotation and annotations that do
not split single exons into multiple entries are ideal, (3) Bedtools is
in your $PATH, and (4) kerneltree is properly installed (you may need to
install Cython first). You may also want to refer to the installation
requirements and/or use the conda
environment for flair.
Usage:
python flair.py correct -q query.bed12 -g genome.fa [options]
run with --help for description of optional arguments. Outputs (1)
bed12 of corrected reads, (2) bed12 of reads that weren’t able
to be corrected, (3) psl of corrected reads if the -c chromsizes
file is provided. Either (1) or (3) can be supplied to flair-collapse as
the query.
Short-read junctions¶
To use short-read splice sites to aid with correction, one option is
bin/junctions_from_sam.py to extract splice junctions from
short-read alignments. The -s option accepts either sam or
bam files, and if there are multiple sams/bams they can be provided
in a comma-separated list.
Usage:
python junctions_from_sam.py -s <shortreads.sam>|<shortreads.bam> -n outname
The file that can be supplied to flair-correct with -j is in the
output file outname_junctions.bed. It is recommended that the user
remove infrequently used junctions i.e. junctions with few supporting
junction reads, which are in the 5th column of the junction bed file.
For example, if you wanted to do the filter out junctions with fewer
than 3 supporting short reads, you could use
awk '{ if ($5 > 2) { print } }' outname.sj_junctions.bed > outname.filtered.bed.
Alternatively, the -j argument for flair-correct can also be
generated using STAR. STAR 2-pass alignment of short reads automatically
produces a compatible splice junction file (SJ.out.tab). We
recommend filtering out junctions with few uniquely mapping reads
(column 7).
flair collapse¶
Defines high-confidence isoforms from corrected reads. As FLAIR does not
use annotations to collapse isoforms, FLAIR will pick the name of a read
that shares the same splice junction chain as the isoform to be the
isoform name. It is recommended to still provide an annotation with
-f, which is used to rename FLAIR isoforms that match isoforms in
existing annotation according to the transcript_id field in the gtf.
Intermediate files generated by this step are removed by default, but
can be retained for debugging purposes by supplying the argument
--keep_intermediate and optionally supplying a directory to keep
those files with --temp_dir.
If there are multiple samples to be compared, the flair-corrected read
bed or psl files should be concatenated prior to running
flair-collapse. In addition, all raw read fastq/fasta files should
either be specified after -r with space/comma separators or
concatenated into a single file.
Usage:
python flair.py collapse -g genome.fa -r <reads.fq>|<reads.fa> -q <query.psl>|<query.bed> [options]
run with --help for description of optional arguments.
Outputs the high-confidence isoforms in several formats: (1)
*isoforms.psl or .bed, (2) *isoforms.gtf, as well as (3) an
*isoforms.fa file of isoform sequences. If an annotation file is
provided, the isoforms ID format will contain the transcript id,
underscore, and then the gene id, so it would look like ENST*_ENSG*
if you’re working with the GENCODE human annotation. If multiple
TSSs/TESs are allowed (toggle with --max_ends or
--no_redundant), then a -1 or higher will be appended to the end
of the isoform name for the isoforms that have identical splice junction
chains and differ only by their TSS/TES. For the gene field, the gene
that is assigned to the isoform is based on whichever annotated gene has
the greatest number of splice junctions shared with the isoform. If
there are no genes in the annotation which can be assigned to the
isoform, a genomic coordinate is used (e.g. chr*:100000).
flair quantify¶
Convenience function to quantifying FLAIR isoform usage across samples
using minimap2. If isoform quantification in TPM is desired, please use
the --tpm option. If the user prefers
salmon to
quantify transcripts using their nanopore reads, please specify a path
to salmon using --salmon. For all options run flair-quantify with
--help.
Usage:
python flair.py quantify -r reads_manifest.tsv -i isoforms.fasta [options]
Inputs:
1. reads_manifest.tsv is a tab-delimited file
containing the sample name, condition, batch*, and path to reads.fq/fa.
For example:
sample1 conditionA batch1 ./sample1_reads.fq
sample2 conditionA batch1 ./sample2_reads.fq
sample3 conditionA batch2 ./sample3_reads.fq
sample4 conditionB batch1 ./sample4_reads.fq
sample5 conditionB batch1 ./sample5_reads.fq
sample6 conditionB batch2 ./sample6_reads.fq
* The batch descriptor is used in the downstream flair-diffExp analysis
to model unintended variability due to secondary factors such as batch
or sequencing replicate. If unsure about this option, leave this column
defined as batch1 for all samples.
2. isoforms.fasta contains FLAIR collapsed isoforms produced by the
flair collapse module.
Outputs: (1) counts_matrix.tsv which is a tab-delimited file
containing isoform counts for each sample. In the output, the values in
the manifest file are concatenated with underscores so please do not use
underscores in the manifest file. For example:
ids samp1_conditionA_batch1 samp2_conditionA_batch1 samp3_conditionA_batch2 ...
0042c9e7-b993_ENSG00000131368.3 237.0 156.0 165.0 150.0 ...
0042d216-6b08_ENSG00000101940.13 32.0 14.0 25.0 ...
flair diffExp¶
Performs differential isoform expression, differential gene expression, and differential isoform usage analyses between multiple samples with 3 or more replicates. For differential isoform usage analysis between samples without replicates, please use the diff_iso_usage.py standalone script. This module requires additional python modules and R packages which are described below:
Additional Requirements¶
Usage:
python flair.py diffExp -q counts_matrix.tsv -o output_directory [options]
Inputs: (1) counts_matrix.tsv is a tab-delimited file
generated by the flair quantify module.
Outputs: (1) Files contained in the output_directory are
tables and plots generated from the various R-packages used in this
analysis, including raw DESeq2/DRIMSeq output tables with foldChange,
isoform frequency and adjusted pvalues.
flair diffSplice¶
Calls alternative splicing events from isoforms. Currently we support the following AS events: intron retention, alternative 3’ splicing, alternative 5’ splicing, and cassette exons.
Usage:
python flair.py diffSplice -i <isoforms.bed>|<isoforms.psl> -q counts_matrix.tsv [options]
If there are 3 or more samples per condition, then the user can run with
--test and DRIMSeq will be used to calculate differential usage of
the alternative splicing events between two conditions. Run with
--help to see more DRIMSeq-specific arguments. If conditions were
sequenced without replicates, then the flair-diffsplice output files can
be input to the diffsplice_fishers_exact.py
script for statistical testing instead.
Inputs: (1) -i is a tab-delimited isoforms.bed/psl file
generated by the `flair-collapse flair collapse module.
(2) -q is a tab-delimited counts_matrix.tsv file generated by the
flair quantify module.
Outputs: (1-4) 4 tab-delimited files for each AS event type. If DRIMSeq was run, and a file with PSIs for each sample and the corresponding p-values for each event type (5-8).
For a complex splicing example, please note the 2 alternative 3’ SS, 3 intron retention, and 4 exon skipping events in the following set of isoforms that flair-diffSplice would call and the isoforms that are considered to include or exclude the each event:
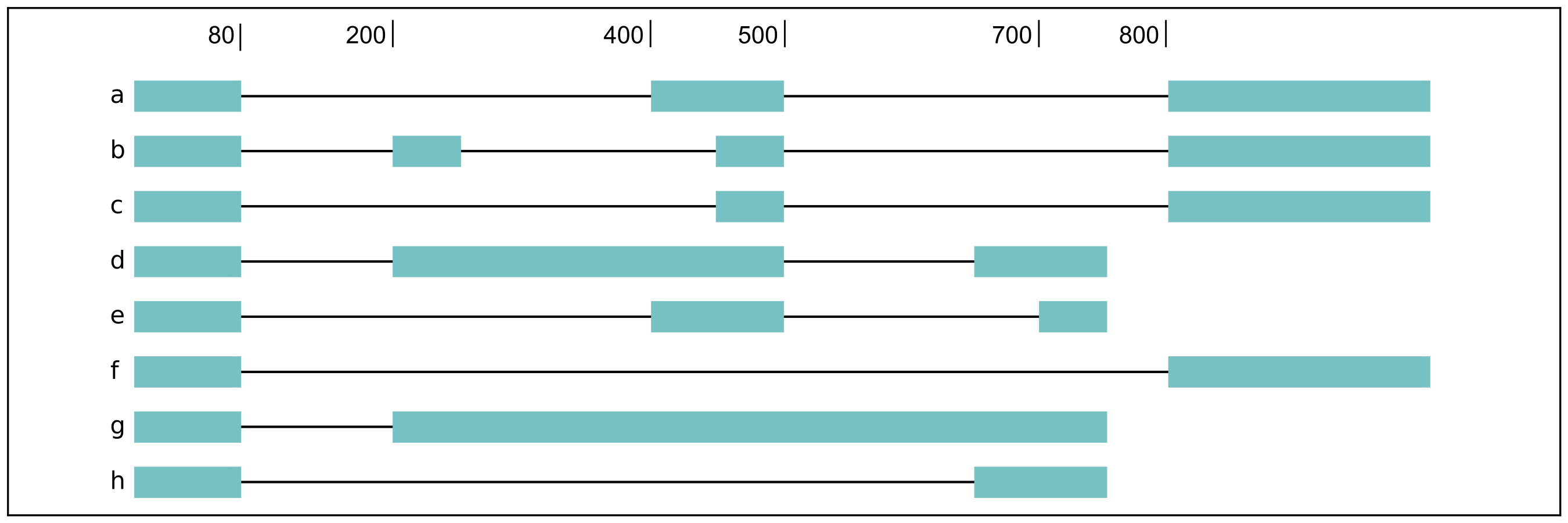
a3ss_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:80 chr1:80-400_chr1:80-450 75.0 35.0 ... a,e
exclusion_chr1:80 chr1:80-400_chr1:80-450 3.0 13.0 ... c
inclusion_chr1:500 chr1:500-650_chr1:500-700 4.0 18.0 ... d
exclusion_chr1:500 chr1:500-650_chr1:500-700 70.0 17.0 ... e
ir_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:500-650 chr1:500-650 46.0 13.0 ... g
exclusion_chr1:500-650 chr1:500-650 4.0 18.0 ... d
inclusion_chr1:500-700 chr1:500-700 46.0 13.0 ... g
exclusion_chr1:500-700 chr1:500-700 70.0 17.0 ... e
inclusion_chr1:250-450 chr1:250-450 50.0 31.0 ... d,g
exclusion_chr1:250-450 chr1:250-450 80.0 17.0 ... b
es_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:450-500 chr1:450-500 83.0 30.0 ... b,c
exclusion_chr1:450-500 chr1:450-500 56.0 15.0 ... f
inclusion_chr1:200-250 chr1:200-250 80.0 17.0 ... b
exclusion_chr1:200-250 chr1:200-250 3.0 13.0 ... c
inclusion_chr1:200-500 chr1:200-500 4.0 18.0 ... d
exclusion_chr1:200-500 chr1:200-500 22.0 15.0 ... h
inclusion_chr1:400-500 chr1:400-500 75.0 35.0 ... e,a
exclusion_chr1:400-500 chr1:400-500 56.0 15.0 ... f